

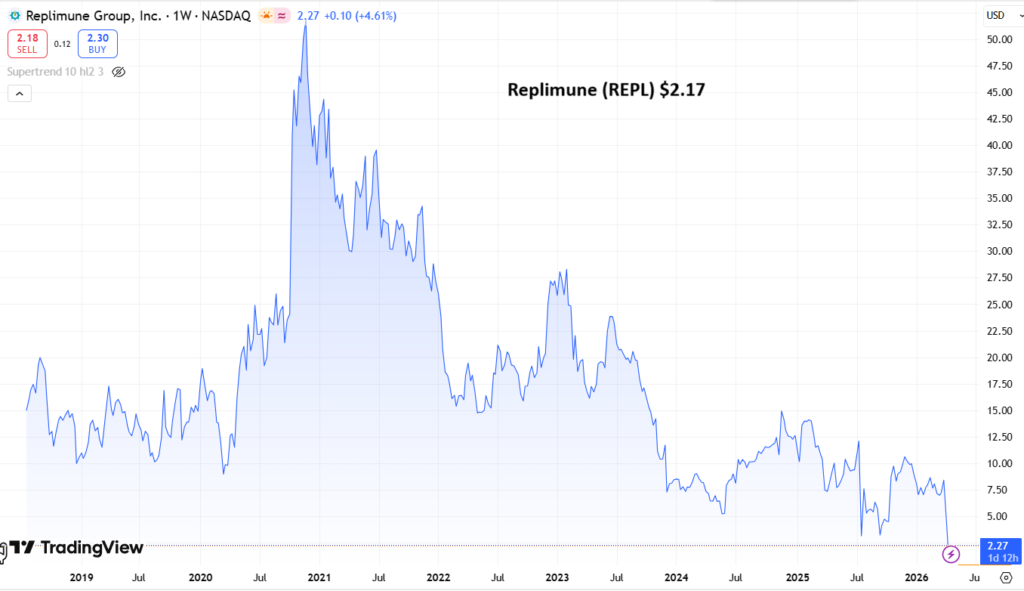
Adding Replimune to 2026 Watch List @ $2.17
Wife: “Honey, I’m going to the mall, do we have money in the checking?” CEO “Yes, but don’t go crazy, we’re down to $269 million and that has to last or we’ll be out of money by next Easter.”
Short (not short-selling) story here is that Replimune (REPL) received the dreaded CRL (Complete Response Letter) from the FDA. We like to call it the TFP-PCBA letter (Thanks For Playing – Please Come Back Again).
What are the odds this bounces higher in the coming month – possibly a double? Never tell me the odds.
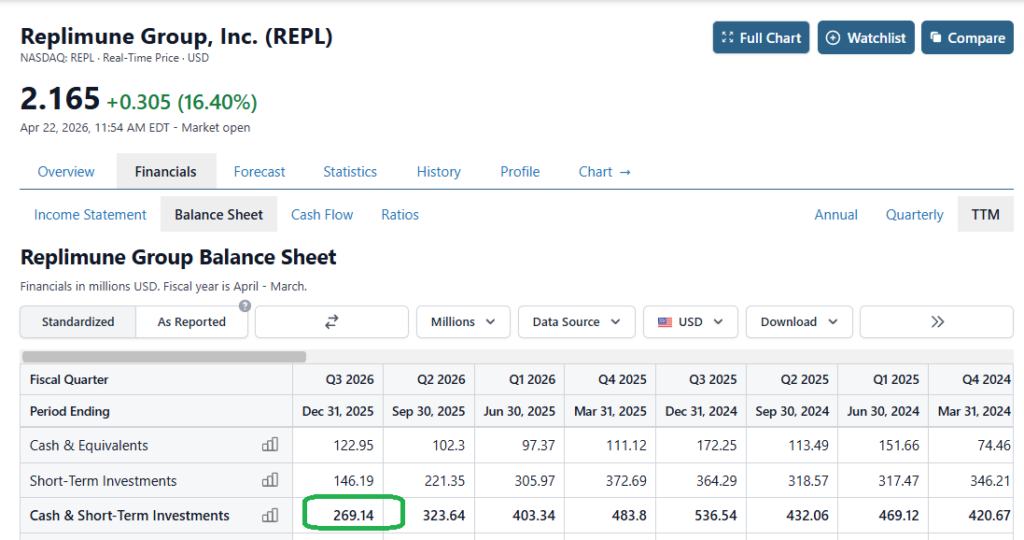
Here’s the balance sheet.. Imagine being the CEO.
- Seeking Alpha Article (bearish). Second CRL May Have Sealed RP1’s Fate – It’s Hard To See Positives
- Wall Street Journal Article (hmmm). RFK Jr. Dodges the Oncologist Heat
3. Replimune Analysts Turn Bearish Following Regulatory Setback
1. Second CRL May Have Sealed RP1’s Fate – It’s Hard To See Positives
Edmund Ingham Apr 10, 2026, 3:30 PM Replimune Group, Inc. (REPL) Stock.
Share Price $4.76 (now $2.17)
Summary
Replimune Group, Inc. suffered a second FDA rejection for RP1 in anti-PD-1 failed melanoma, causing a sharp stock decline and casting doubt on its near-term prospects.
The FDA apparently cited inadequate evidence of effectiveness and trial design flaws, finding the IGNYTE study too heterogeneous and the confirmatory study insufficient.
REPL’s cash runway may not extend beyond 12 months, and the company may need to restructure or reprioritize resources, with RP2 in metastatic uveal melanoma as a possible focus.
Given persistent regulatory hurdles and uncertain funding, this does not appear to be a “buy the dip” opportunity for REPL.
News is breaking today that Replimune Group, Inc.’s (REPL) Biologics License Application (“BLA”) requesting approval for its drug candidate RP1 (vusolimogene oderparepvec) in the indication of anti-PD-1 failed melanoma has been rejected by the FDA – the second time the FDA has rejected the drug for commercial approval.
At the time of writing, Replimune stock has fallen in value by nearly 20%, trading at a value of $4.76 per share – trading is currently halted. Stock is down >50% year-to-date, and nearly 85% on a five-year basis.
In this note, I’ll provide some background and context to today’s news and speculate about what it means for the company and its share price and valuation going forward.
Investment Overview – Background To Today’s Decision
This is in fact my first time covering Replimune Group for Seeking Alpha, although I have frequently referenced the Woburn, Massachusetts-based biotech in my coverage of Iovance Biotherapeutics (IOVA) in the past.
As many readers will likely be aware, and as I noted in a March update on Iovance:
Iovance’s lifileucel, brand name Amtagvi, is a commercially available therapy for advanced melanoma, having been approved in February 2024, and together with Proleukin (aldesleukin), an interleukin-2, or IL-2, product used in the Amtagvi treatment regimen, helped Iovance post a revenue figure of $264m in 2025.
it took far longer than expected to secure approval for Amtagvi than initially thought – the original biologics license application (“BLA”) requesting FDA approval of the drug was submitted in 2022, but approval was not granted until 2024, testing Wall Street’s patience.
Replimune’s lead product is RP1, indicated for anti-PD-1 failed melanoma, i.e., patients did not respond to treatment with an immune checkpoint inhibitor – likely Merck’s (MRK) Keytruda (pembrolizumab), or Bristol-Myers Squibb’s (BMY) Opdivo (nivolumab).
Replimune, like Iovance, has undergone a long and tortuous path towards an FDA approval, however its journey has ultimately not been completed – as per its fiscal year Q3 10-Q filing from early February:
In November 2024, we announced submission of our first Biologics License Application, or BLA, to the U.S. Food and Drug Administration, or the FDA, for RP1 (vusolimogene oderparepvec) in combination with nivolumab for the treatment of adult patients with advanced melanoma who have previously received an anti-PD-1 containing regimen, and that the FDA has granted Breakthrough Therapy designation for RP1 in combination with nivolumab in the same setting.
The submission was made under the accelerated approval pathway. The FDA accepted our BLA and granted priority review with a Prescription Drug User Fee Act, or PDUFA, goal date of July 22, 2025. On July 21, 2025 the FDA issued a complete response letter, or CRL, for the RP1 BLA for the treatment of advanced melanoma.
The dreaded CRL outlined the FDA’s reasons for rejecting a application to approve a drug. Iovance notes:
The FDA stated in the CRL that it was unable to approve the application in its present form and that the IGNYTE trial was not considered to be an adequate and well-controlled clinical investigation that provided substantial evidence of effectiveness, including contribution of components. Furthermore, the FDA said the trial could not be adequately interpreted due to the heterogeneity of the patient population.
Clearly, Replimune was confident that it had addressed the FDA’s concerns – during a fireside chat at this year’s JP Morgan Healthcare conference in January, its CEO had told the audience:
“So near term, we’re looking forward to our approval, and we’re already ready to execute on our commercial launch plans. That involves working very closely with the oncology and interventional radiology stakeholders. We’ve done a lot of work on now and ready to go. And we’re also worked to simplify the logistics for our modalities so that we can enable next-day delivery and also stability at room temperature for our asset.”
In its press release announcing the FDA acceptance of its resubmitted BLA, Replimune describes the mechanism of action (“MoA”) of RP1 as follows:
RP1 (vusolimogene oderparepvec) is Replimune’s lead product candidate and is based on a proprietary strain of herpes simplex virus engineered and genetically armed with a fusogenic protein (GALV-GP R-) and GM-CSF, intended to maximize tumor killing potency, the immunogenicity of tumor cell death, and the activation of a systemic anti-tumor immune response.
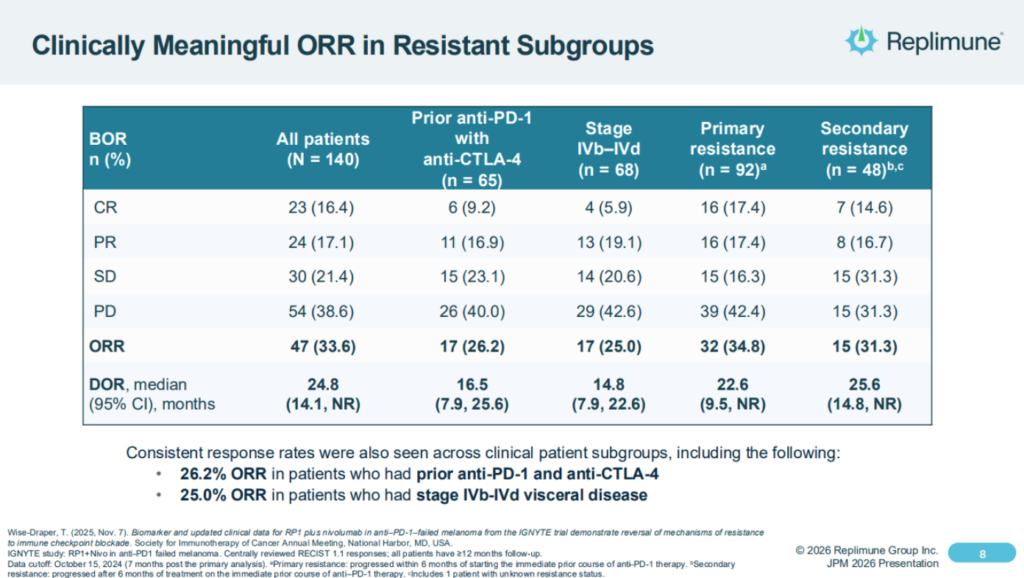
It’s pivotal study used RP1 in combo with nivolumab – Bristol-Myers Squibb’s (BMY) PD-1 inhibitor Opdivo, and achieved an objective response rate of ~33%, however the FDA apparently concluded the study has enrolled too broad a patient population, making it hard to assess results, and was also unhappy with the design of a “confirmatory” study that Replimune intended to conduct post-approval, to confirm its Phase 1/2 data.
Replimune shares the following overview of IGNYTE study results as follows in its latest investor presentation:
As the CEO explained back in January, the company had confidence that it was RP1, not the previously administered PD-1 therapy that was doing the “heavy lifting:
So one of the important questions you always get when you’re doing a single-arm trial, particularly with the combination is well, what is RP1 contributing? And I mentioned one of the ways we got to that was this very strict criteria so that we know that further PD-1-based treatments really wouldn’t do very much, as I said, about 5%. So when you see about a 34% response rate, we know that the RP1 is really contributing.
At the time of the first rejection, there was widespread surprise at the FDA’s rejection, with some blaming the new regime at the FDA – notably, Dr Vinay Prasad, the former Director of the Center for Biologics Evaluation and Research (“CBER”). With Prasad set to leave the FDA at the end of this month, however, it seems the FDA remains broadly unconvinced by Replimune’s data set, and does not believe the updated data and analytics provided since a Type A meeting last year materially changes the case for approval.
The reality is, oncolytic viruses have struggled to prove their mettle both in the clinic (RP1 formerly failed a study in cutaneous squamous cell carcinoma), and commercially – Amgen’s Imlygic, approved to treat melanoma in 2015, likely earns <$100m per annum (Amgen does not break out its revenue contribution but would likely do so if the figure was >$100m).
Replimune – What Happens Now?
Announcing its fiscal Q3 earnings in February this year, Replimune reported a net loss of $(247m), up from $(182m) in the prior year period, and a cash position of $269m.
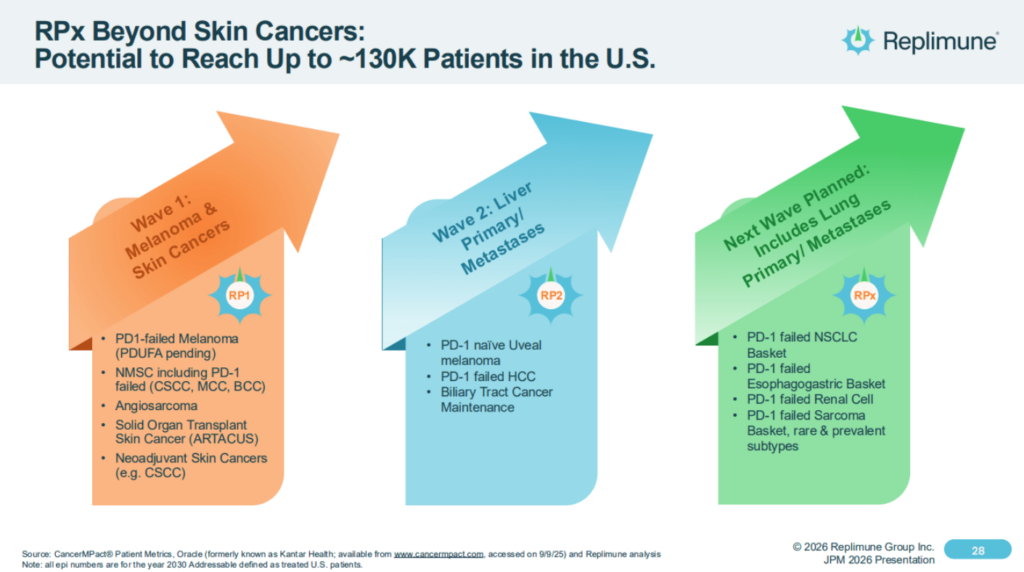
Replimune’s plans have always extended beyond approval in advanced melanoma to indications addressing ~130k cancer patients, as illustrated in the slide below:
Doubtless, Replimune management will feel hard done by today and will want to press ahead with studies, but seemingly, with a cash position of $269.1M and a quarterly cash burn of $70.9M, its funding runway may only extend into the first quarter of 2027. It’s likely Replimune would have looked to raise funds via a stock offering if its BLA had been approved today, at a much higher price than the current share price value of <$5, to fund a commercial launch, but it may now be challenging for the company to secure funding, or even issue debt.
In terms of the objective response rates (“ORRs”) and Complete Response Rates (“CRs”) observed in the IGNYTE study, they do seem broadly comparable to Amtagvi. However, Amtagvi seems to have performed well in the commercial setting – five-year analysis of the pivotal study of Amtagvi in melanoma patients revealed an ORR of 31.4%, a five-year overall survival (“OS”) of 19.7%, a median duration of response (“mDor”) of 36.5 months, and a median OS of 13.9 months.
Ultimately, however clinical studies are rarely comparable, patients have contrasting profiles, may respond differently to a particular therapy, and trial designs and outcomes must be crystal clear and pre-agreed with the regulatory authorities. Rightly or wrongly, the FDA was seemingly convinced by Iovance’s data submission, and unconvinced by Replimune’s.
At the time of writing, Replimune has not released any communications round the FDA’s decision – management will doubtless believe they had reached agreement with the administration on the data required to secure approval, and supplied it, but the final decision always rests with the administration (for better or worse).
April 10 Release (post report)
“It is deeply disappointing that the FDA has not exercised regulatory flexibility to meet patients’ needs given the data supporting strong efficacy and the favorable safety profile. Approximately 8,500 Americans with advanced melanoma die every year. The country’s foremost melanoma specialists stood behind the RP1 data. Patients and caregivers pleaded for urgency. All of it was met with inconsistent communication and a fragmented and slow-moving regulatory process which clearly puts U.S. innovation at risk,” said Sushil Patel, Ph.D., CEO of Replimune. “As we previously communicated, without timely accelerated approval, the development of RP1 will not be viable. We are devastated for our committed employees who have worked tirelessly for patients but at this point we have no choice but to eliminate jobs, including substantially scaling back our U.S. based manufacturing operations. A treatment desperately needed by patients will not be available. Not because the medicine failed. Because the system did.”
Dems fighting words!
As such, it is hard to know how the company will react. The company’s market cap has sunk to <$400m, and it may be the case Replimune will need to scale back operations and reprioritize resources – it would not necessarily be surprising to see a corporate restructuring, in order to reduce costs.
The post-confirmatory study in advanced melanoma is surely no longer required, and in many senses it feels as though Replimune is back at square one. The company does have a second program, RP2, currently progressing through a Phase 2/3 study in metastatic uveal melanoma, and I wonder if management will opt to prioritize this over RP1 going forward. Management says that:
The primary endpoints of the trial are overall survival and progression free survival, and key secondary endpoints are overall response rate and disease control rate. Phase 2/3 transition is expected in Q1 2027, with PFS analysis potentially supporting accelerated approval.
Concluding Thoughts – A Tough Day For Replimune, A Quick Turnaround In Fortunes Feels Unlikely
My take on today’s news is that the FDA has doubled down on the criticism of Replimune’s pivotal study it shared with its first CRL, and does not believe its criticisms have been addressed by the re-submission.
Facing its second rejection, my take would also be that Replimune cannot have too many complaints. Oncolytic viruses, while a promising potential therapy, have defied the best efforts of several pharmas and biotechs to devise an effective and safe therapy, and there did not seem to be enough signs that RP1 would have been a drastic improvement on Amtagvi, or potentially even PD-1 plus chemotherapy, while other melanoma therapies – Moderna’s (MRNA) / Merck’s (MRK) cancer “Vaccine” candidate for example, may soon provide another treatment option in a field of admittedly high unmet need.
For Replimune, its feels as though it is back to the drawing board, and management may need to take some tough decisions. It will certainly be interesting to see the company’s response, but whatever it may be, personally, and doubtless many readers will disagree, this does not feel like a “buy the dip” moment for Replimune stock.

2. Wall Street Journal Article (hmmm). RFK Jr. Dodges the Oncologist Heat
The HHS Secretary tells the Senate he didn’t kill a melanoma drug.
The Food and Drug Administration rejection of Replimune’s life-saving treatment for metastatic melanoma is so dubious that even Health and Human Services Secretary Robert F. Kennedy Jr. is distancing himself from it.
During a hearing Wednesday, Nevada Sen. Catherine Cortez Masto asked Mr. Kennedy about our recent editorial that noted criticisms from oncologists about the FDA rejection of Replimune’s RP1 immunotherapy. “First of all, I had nothing to do with this decision,” he replied.
“I’ve been told by [FDA Commissioner] Marty Makary that every panel that looked at that drug unanimously voted against it,” he said. If so, he was misled. As we wrote, the first FDA panel to review the drug supported its approval but was overruled by biologics chief Vinay Prasad. We’re also told that staffers who recommended approval were sidelined. Retaliation?
Mr. Kennedy said he was told FDA staff “voted against it because it does not appear to work, because the trial was a one-armed trial when the company was asked to do a two-armed trial.” That’s more misinformation. As oncologists explained to us and in letters to the FDA, it would have been unethical to do a trial with patients in a control arm receiving a drug on which their cancers would get worse.
FDA staff in 2024 designated RP1 a “breakthrough” therapy because they were so impressed by its trial results. One-third of patients who failed to respond to other immunotherapies went into remission, and tumors shrank in nearly all patients.
Ms. Cortez Masto pressed: “Why would you prevent the opportunity for lifesaving drugs in individuals?”
Mr. Kennedy: “That’s a great question. Why would I? Why would I?”
Ms. Cortez Masto: “That’s exactly right. So I’m going to ask you to take a second look at it.”
Mr. Kennedy seemed to agree: “If the drug works, we’re going to approve it.”
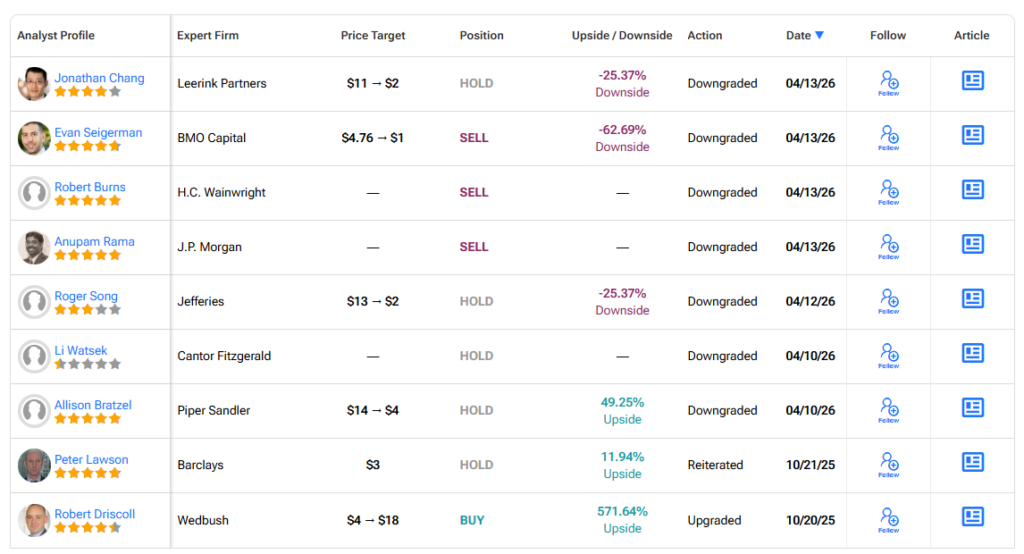
3. Replimune Analysts Turn Bearish Following Regulatory Setback
HC Wainwright downgraded Replimmune from Buy to Sell. Analyst Robert Burns sees a significantly longer path for RP-1 to reach the market, if at all.
“It may take until late in this decade to furnish the agency with the data that it appears to be demanding. There can be no assurance that Replimune would be able to accomplish this. As such, the company is facing a significant crisis and may not be able to pivot successfully,” Burns added.
PRESS RELEASE
Replimune Receives Complete Response Letter from the FDA for RP1 Biologics License Application for the Treatment of Advanced Melanoma | April 10, 2026 17:02 ET
April 10, 2026 17:02 ET
WOBURN, Mass., April 10, 2026 (GLOBE NEWSWIRE) — Replimune Group, Inc. (NASDAQ: REPL), a clinical stage biotechnology company pioneering the development of novel oncolytic immunotherapies, today announced that the company received a complete response letter (CRL) from the U.S. Food and Drug Administration (FDA) for the Company’s Biologics License Application (BLA) for RP1 in combination with nivolumab for the treatment of advanced melanoma.
Replimune disagrees with the FDA about whether the data set, upon which breakthrough therapy designation was awarded, is sufficient to allow this promising medicine to be made available to advanced cancer patients. In the IGNYTE trial, patients with confirmed progression on an anti-PD-1 based regimen who received RP1 plus nivolumab had a 34% response rate with a median duration of 24.8 months with a favorable safety profile.
“It is deeply disappointing that the FDA has not exercised regulatory flexibility to meet patients’ needs given the data supporting strong efficacy and the favorable safety profile. Approximately 8,500 Americans with advanced melanoma die every year. The country’s foremost melanoma specialists stood behind the RP1 data. Patients and caregivers pleaded for urgency. All of it was met with inconsistent communication and a fragmented and slow-moving regulatory process which clearly puts U.S. innovation at risk,” said Sushil Patel, Ph.D., CEO of Replimune. “As we previously communicated, without timely accelerated approval, the development of RP1 will not be viable. We are devastated for our committed employees who have worked tirelessly for patients but at this point we have no choice but to eliminate jobs, including substantially scaling back our U.S. based manufacturing operations. A treatment desperately needed by patients will not be available. Not because the medicine failed. Because the system did.”
Inconsistent agency process and communication thwarts innovation
With the CRL, the company learned that a different review team was appointed for the resubmission and replaced the prior team who had interacted with the company. A senior member of the prior review team stated publicly that the “BLA clinical team thought the applicant had provided adequate evidence to support contribution of effect of RP1 plus nivolumab but leadership did not agree.” The new team did not meet with the company during the review process despite the company offering.
In the CRL, the agency appears to have contradicted their positions expressed at the September 2025 Type A meeting, including on the following points:
- After testimony from melanoma experts, the agency did not raise further concerns about the heterogeneity of the patient population in IGNYTE and acknowledged that randomizing patients to an anti-PD1 only arm in the confirmatory study was not feasible.
- Following an agency suggestion, the company submitted a proposal for a descriptive analysis from IGNYTE-3 supporting contribution of components. The company also included data from IGNYTE showing median progression free survival on RP1 plus nivolumab was 30.6 months in responding patients compared to 4.4 months on their prior PD-1 based regimen. The company requested feedback, however, the FDA did not respond and subsequently accepted the resubmission as a complete response to the July 2025 CRL.
- The FDA raised several points related to tumor assessment methodology. As requested by the FDA, responses in IGNYTE were assessed using RECIST 1.1 without modifications. In addition, the company provided detailed analyses showing no material difference in response rates between injected and non-injected lesions. The company also provided a comprehensive analysis which showed that biopsies and surgical interventions did not impact tumor response.
Prior to the original BLA submission, standard regulatory meetings were conducted to discuss trial design, patient population, and the BLA package requirements. While a randomized controlled trial was preferred, the FDA suggested in the March 2021 Type B minutes that if the data was sufficiently compelling, a single arm trial could be acceptable for consideration under accelerated approval. At the subsequent pre-BLA meeting, the FDA stated “we do not object to your proposal to submit a BLA based primarily on data from the cohort of patients (n=140) in the Phase 2 IGNYTE trial who had advanced melanoma and progressed while being treated with prior anti-PD-1 based therapy.” The company subsequently submitted a BLA which was accepted with breakthrough therapy designation and granted priority review. Based on feedback from the FDA, the company initiated a resource-intensive global Phase 3 trial, IGNYTE-3, to satisfy the regulatory requirement that a confirmatory study be underway for an accelerated approval.
About Melanoma
Melanoma is the fifth most common cancer, with approximately 112,000 new cases estimated in the U.S. in 2026, and the most lethal form of skin cancer, accounting for nearly 8,500 deaths annually. Standard of care therapy includes treatment with immune checkpoint blockade, to which approximately half of patients will not respond or will progress after treatment. Melanoma is considered advanced when the cancer spreads beyond the primary tumor to other parts of the body.
About RP1
RP1 (vusolimogene oderparepvec) is Replimune’s lead product candidate and is based on a proprietary strain of herpes simplex virus engineered and genetically armed with a fusogenic protein (GALV-GP R-) and GM-CSF intended to maximize tumor killing potency, the immunogenicity of tumor cell death, and the activation of a systemic anti-tumor immune response.
About Replimune
Replimune Group, Inc., headquartered in Woburn, MA, was founded in 2015 with the mission to transform cancer treatment by pioneering the development of novel oncolytic immunotherapies. Replimune’s proprietary RPx platform is based on a potent HSV-1 backbone intended to maximize immunogenic cell death and the induction of a systemic anti-tumor immune response. The RPx platform is intended to ignite local activity consisting of direct selective virus-mediated killing of the tumor resulting in the release of tumor derived antigens and altering of the tumor microenvironment to then activate a strong and durable systemic response. The RPx product candidates are expected to be synergistic with most established and experimental cancer treatment modalities, leading to the versatility to be developed alone or combined with a variety of other treatment options. For more information, please visit www.replimune.com.
Forward Looking Statements
This press release contains forward looking statements within the meaning of Section 27A of the Securities Act of 1933, as amended, and Section 21E of the Securities Exchange Act of 1934, as amended, including statements regarding our interactions with the FDA and other statements identified by words such as “could,” “expects,” “intends,” “hope,” “may,” “plans,” “potential,” “should,” “will,” “would,” or similar expressions and the negatives of those terms. Forward-looking statements are not promises or guarantees of future performance, and are subject to a variety of risks and uncertainties, many of which are beyond our control, and which could cause actual results to differ materially from those contemplated in such forward-looking statements. These factors include risks related to our limited operating history, our ability to generate positive clinical trial results for our product candidates, the costs and timing of operating our in-house manufacturing facility, the timing and scope of regulatory approvals, if any, our ability to resolve the issues identified in the CRL in a manner satisfactory to the FDA and to us and the timing thereof, the availability of combination therapies needed to conduct our clinical trials, changes in laws and regulations to which we are subject, competitive pressures, our ability to identify additional product candidates, political and global macro factors including the impact of a global pandemic and related public health issues and the ongoing political and military conflicts, including trade conflicts, and other risks as may be detailed from time to time in our Annual Reports on Form 10-K and Quarterly Reports on Form 10-Q and other reports we file with the Securities and Exchange Commission. Our actual results could differ materially from the results described in or implied by such forward-looking statements. Forward-looking statements speak only as of the date hereof, and, except as required by law, we undertake no obligation to update or revise these forward-looking statements.
Investor Inquiries
Chris Brinzey
ICR Healthcare
339.970.2843
chris.brinzey@icrhealthcare.com
Media Inquiries
Arleen Goldenberg
Replimune
917.548.1582
media@replimune.com
 $0.27, Up 42%, to Watch List.")
. Palantir’s Open Secure AI.")


 $7.22. We’re Up And Out, With a 301% Gain.")

 $0.27, Up 42%, to Watch List.")
. Palantir’s Open Secure AI.")
 Jumps 100%.")
{kind=link}